
CE Marking for Medical Devices
EU MDR2017/745
CE Marking คือเครื่องหมายแสดงถึงประสิทธิภาพและความปลอดภัยสำหรับผลิตภัณฑ์ที่ต้องการนำเข้าวางจัดจำหน่ายในสหภาพยุโรป ตัวอย่างกฎระเบียบข้อบังคับผลิตภัณฑ์ที่ต้องมีการรับรอง CE Mark ก่อนได้รับอนุญาตและจัดจำหน่าย ซึ่งมีมากกว่า 20 กลุ่มผลิตภัณฑ์ที่ต้องได้รับรองมาร์ค (Product groups)
- PPE 2106/425 : Personal protective equipment
- Directive 2006/42/EC : Machinery
- Directive 2014/35/EU : Low Voltage
- Directive 2014/33/EU : Lifts
- Directive 2014/68/EU : pressure equipment
- Directive 92/42/EEC : Hot-water boilers
ในปัจจุบันสำหรับผลิตภัณฑ์เครื่องมือแพทย์ แบ่งเป็น 2 หมวดหลักคือ
- EU MDR 2017/745 : Medical Devices
- EU IVDR 2017/746 : In-vitro diagnostic medical devices
ดังนั้น ผู้ผลิตและต้องการได้รับการรับรอง CE Mark ต้องตรวจสอบก่อนว่า ผลิตภัณฑ์เครื่องมือแพทย์ อยู่ในกลุ่มใด ตามวัตถุประสงค์การใช้เครื่องมือแพทย์ และในบทความนี้ ขออธิบายถึง EU MDR2017/745 หรือ กลุ่มเครื่องมือแพทย์ที่มีวัตถุประสงค์เพื่อการรักษา บำบัด บรรเทา ฯ (หรือ Non-IVD เครื่องมือแพทย์ที่ไม่ใช่เครื่องมือแพทย์สำหรับการวินิจฉัยภายนอกร่างกาย)
EU MDR2017/745 คือกฎระเบียบที่ได้รับการปรับปรุงและประกาศบังคับใช้ ทั้งนี้เพื่อยกระดับความเชื่อมั่นในผลิตภัณฑ์เครื่องมือแพทย์ที่ต้องมีความปลอดภัย ให้มีความทันสมัยต่อเทคโนโลยี นวัตกรรมของเครื่องมือแพทย์ เช่นการนำโปรแกรม ซอฟท์แวร์ AI หรือปัญญาประดิษฐ์ มาใช้เพื่อประมวลผลหรือรวบรวมข้อมูลด้านสุขภาพของผู้ป่วย กฎระเบียบ MDR ฉบับใหม่นี้จึงมีเป้าหมายเพื่อให้เกิดความโปร่งใสและความสอดคล้องมากขึ้น นอกจากนี้ EU MDR2017/745 จึงเป็นกฎระเบียบที่มาแทน MDD93/42/EEC และ AIMD90/385/EEC
ตัวอย่างประเด็นจาก MDD เป็น MDR
- “implant card” สำหรับผลิตภัณฑ์เครื่องมือแพทย์ที่ฝังในร่างกายผู้ป่วย
- “traceability system” การกำหนดระบบการสอบกลับได้ โดยใช้ระบบฐานข้อมูล EUDAMED และ UDI Label ติดตามย้อนกลับเครื่องมือแพทย์ได้ ตลอดวงจรชีวิตเครื่องมือแพทย์ (Life cycle)
- “Post Market Surveillance” ระบบการติดตามหลังผลิตภัณฑ์วางตลาด
- “Clinical Evidence” การแสดงหลักฐานด้านคลินิกหรือทางการแพทย์ที่เพียงพอที่จะยืนยันด้านความปลอดภัยและประสิทธิภาพของเครื่องมือแพทย์ (Safety and Performance)
- “Supply Chain” การสอบกลับได้ตลอดห่วงโซ่ของเครื่องมือแพทย์ โดยครอบคลุมถึง ผู้ผลิต (Manufacturer) ---ผู้นำเข้า (Importer) -- -ผู้กระจายสินค้า (Distributor) และตัวแทนที่ได้รับการแต่งตั้ง (Authorized Representative) กฎระเบียบยังกำหนดบทบาทหน้าที่ความรับผิดชอบของแต่ละหน่วยงานไว้อย่างชัดเจน
9 Steps เพื่อจัดทำและขอการรับรอง CE Marking
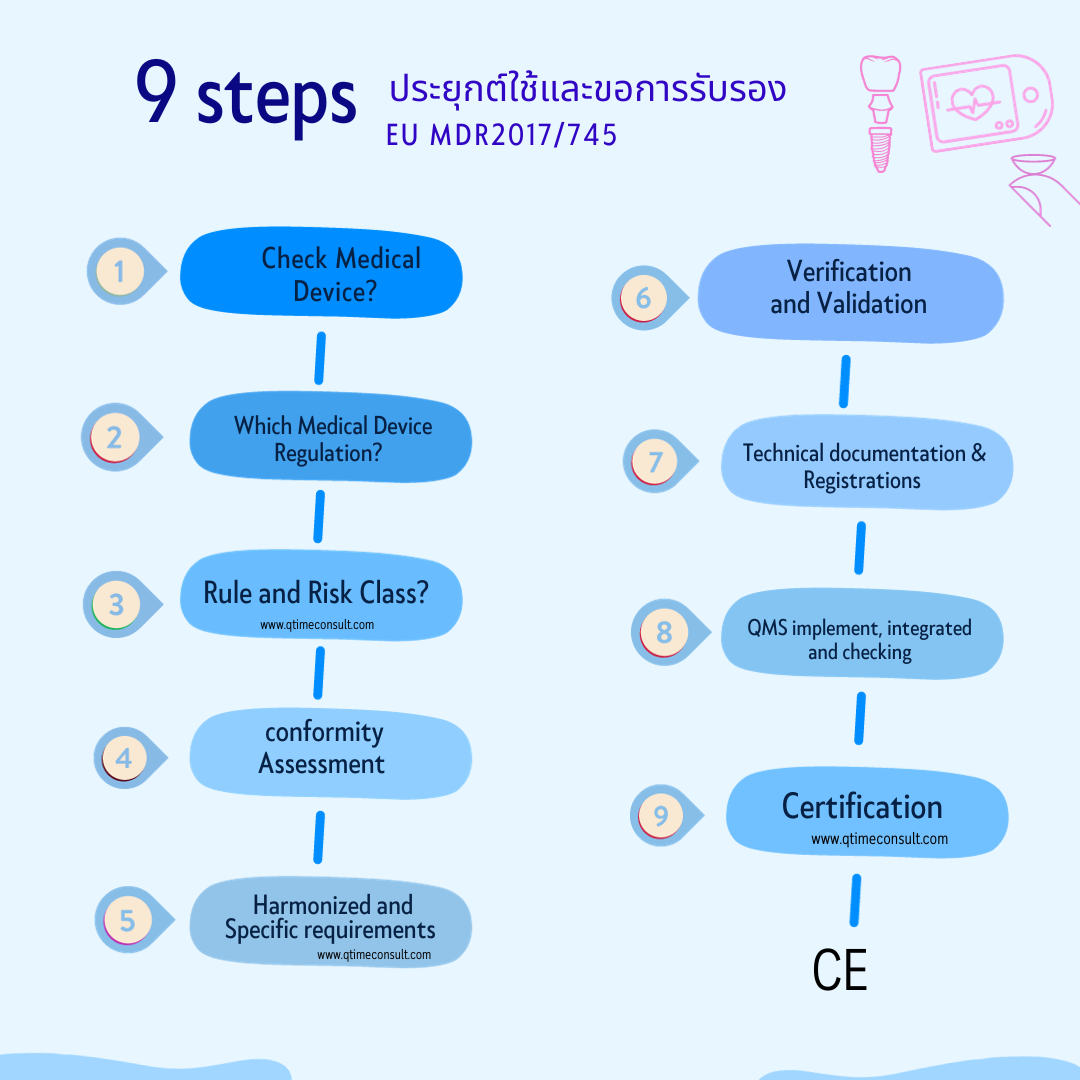
Step 1: ตรวจเช็คผลิตภัณฑ์เป็นเครื่องมือแพทย์ ตามความหมายของ MDR หรือไม่
ผลิตภัณฑ์เครื่องมือแพทย์ ตามความหมายที่ระบุใน MDR2017/745 คือ
The MDR defines (Article 2)
“medical device” means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,
and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means. The following products shall also be deemed to be medical devices:
— devices for the control or support of conception;
— products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
Step 2: จัดเป็นเครื่องมือแพทย์ในกฎระเบียบที่ใด MDR2017/745 หรือ IVDR2017/746
ตามวัตถุประสงค์ และกฎระเบียบ ข้างต้น
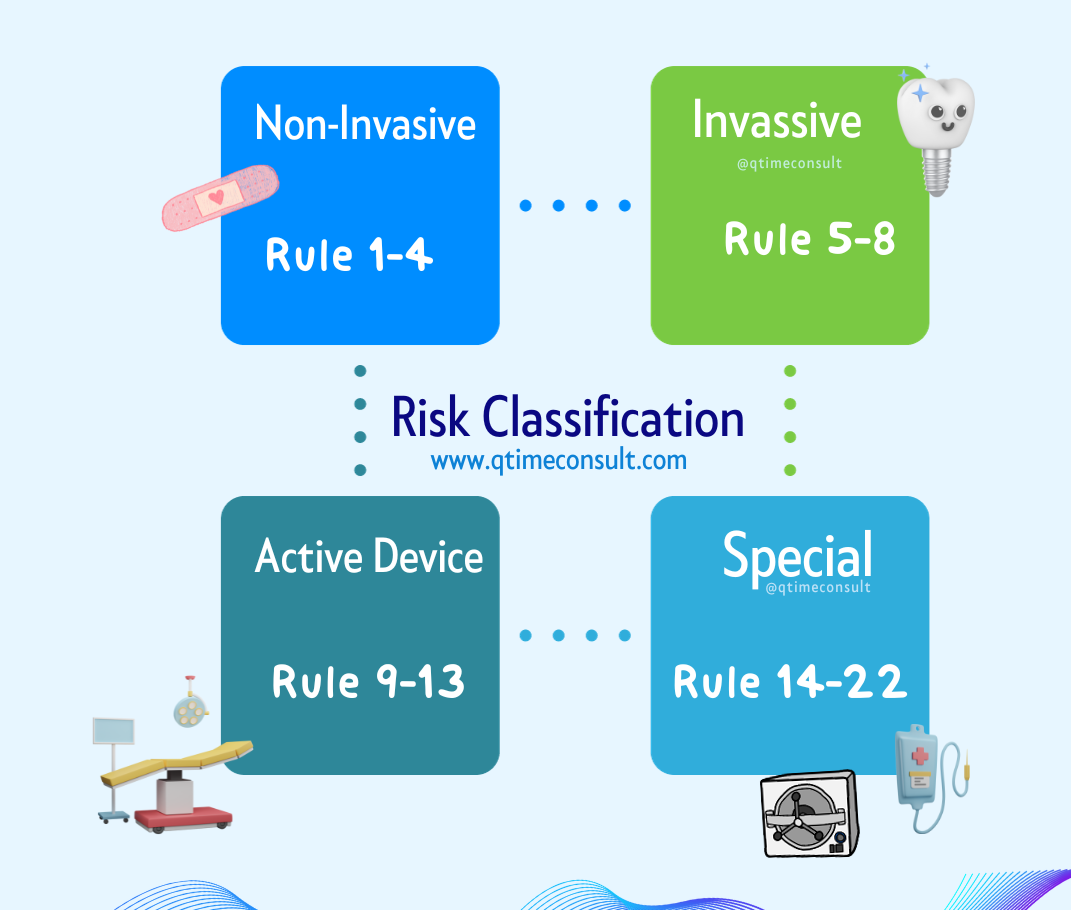
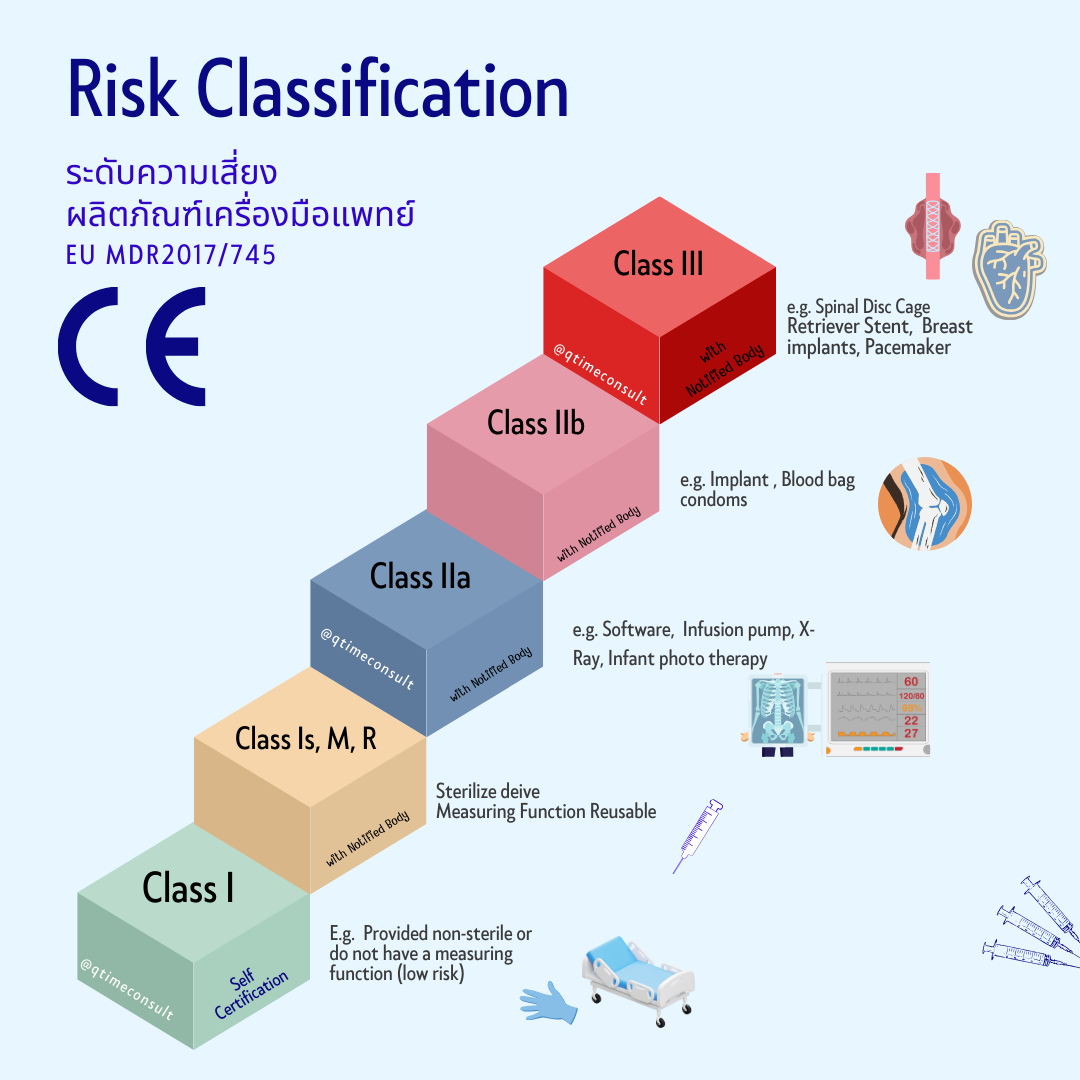
Step 3 : ผลิตภัณฑ์อยู่ในหลักเกณฑ์ (Rule) และระดับความเสี่ยง (Risk classification) ที่เท่าไหร
Step 4 : ตรวจเช็คและพิจารณาแนวทางการยื่นแสดงการรับรองและขอใช้ CE Mark หรือ conformity Assessment ใด
ในกฎระเบียบ MDR ระบุแนวทางวิธีการแสดงการรับรองไว้ คือ
- Annex IV : Annex V
- Annex IX : Conformity Assessment based on QMS and assessment of TF
- Annex X : Type Examination Conformity Assessment
- Annex XI-Part A : Production Quality System Verification
- Annex XI –Part B : Product Verification done by Notified Body
Step 5 : ตรวจสอบมาตรฐาน ข้อกำหนด มาตรฐานที่เกี่ยวข้อง และมาตรฐานผลิตภัณฑ์
(EU Harmonized Standards and product specific requirements)
Sample of Standards ตัวอย่างมาตรฐานและข้อกำหนดที่เกี่ยวข้อง
- ISO 13485:2016 Quality Management System
- ISO14971:2019 Risk Management
- ISO15223-1-2021 Symbols for medical device labelling and information to be supply
- EN1041 Information supplied by the manufacturer of medical devices
- EN ISO 10993-1:2018 Biological evaluation of medical devices
- EN 14683:2019 Medical face masks
- EN 60601-2-12:2006 Requirements for the safety of lung ventilators
- EN ISO 21534:2009 Non-active surgical implants – Joint replacement implants
- EN 455-1:2000 Medical gloves for single use-Freedom from hole
- EN 1618:1997 Catheters other than intravascular catheters
Step 6 : Verification and Validation การทดสอบและตรวจรับรองความถูกต้องของผลิตภัณฑ์ สอดคล้องตาม Harmonized standards หรือ กฎหมาย ข้อกำหนดเฉพาะผลิตภัณฑ์
ทบทวนและตรวจสอบ การทดสอบผลิตภัณฑ์ จากหน่วยงานเช่น ห้องปฏิบัติการที่ได้ผ่านการรับรอง หรือห้องปฏิบัติการที่มีคุณสมบัติที่กำหนด หัวข้อการทดสอบผลิตภัณฑ์
- การทดสอบความปลอดภัยทางไฟฟ้า (Electrical Safety Test)
- การทดสอบการรบกวนทางแม่เหล็กไฟฟ้า (Electromagnetic)
- การทดสอบด้านกายภาพ (Physical Test)
- การทดสอบด้านประสิทธิภาพ (Performance Test)
- การทดสอบด้านชีววิทยา หรือจุลินทรีย์ (Biological Test)
- การทดสอบความเข้ากันได้ทางชีวภาพ (Biocompatibility Test)
- การทดสอบอายุจัดเก็บผลิตภัณฑ์ (Shelf life study)
- การตรวจรับรองความถูกต้องของกระบวนการปราศจากเชื้อ (Sterilization validation)
- การทดสอบความปราศจากเชื้อ (Sterility Test)
- การทดสอบการใช้งานเครื่องมือแพทย์ (Usability Engineering Test)
- และการทดสอบหรือศึกษาประสิทธิภาพและความปลอดภัยเฉพาะอื่นๆ
- การตรวจรับรองความถูกต้องของผลิตภัณฑ์ เช่นข้อมูลด้านคลินิก Clinical Evaluation และ Clinica Investigation
Step 7 : เตรียมจัดทำ Technical Documentation
ข้อมูลผลิตภัณฑ์เครื่องมือแพทย์ที่ต้องการขอการรับรอง ทั้งการออกแบบและพัฒนาผลิตภัณฑ์ รวมถึงการทดสอบและตรวจรับรองความถูกต้องของผลิตภัณฑ์
รายการจัดทำ Technical Documentation File-TDF
- Device Description and Specification / Accessories
- Instruction for use & Labeling
- Design and Manufacturing Information
- General Safety and Performance Requirements (SPR)
- Benefit-Risk analysis
- VERIFICATION AND VALIDATION : Testing and clinical Evidence
- Post Market Surveillance (PMS)
Step 8 : QMS ระบบบริหารคุณภาพ หรือ ISO13485 ตรวจสอบการประยุกต์ใช้โดยต้องครอบคลุมผลิตภัณฑ์ที่ต้องการขอการรับรอง CE Marking ด้วย
- มาตรฐาน ISO13485 คือมาตรฐานพื้นฐานด้านการจัดการระบบบริหารคุณภาพสำหรับผลิตภัณฑ์เครื่องมือแพทย์ และเป็นส่วนหนึ่งของ Harmonized standards ส่วนที่เพิ่มเติมสำหรับผู้ผลิตที่ต้องการขอการรับรอง CE Mark ต้องนำระเบียบข้อกำหนด กฎหมายที่เกี่ยวข้องมาบูรณาการณ์ ร่วมกับระบบ ISO13485 เช่น ระบบการสอบกลับได้ ระบบบริหารห่วงโซ่ (Supply Chain)
ขั้นตอนและแนวทางการประยุกต์ใช้ ISO13485
Step 9 : Certification เมื่อตรวจสอบความพร้อมครบถ้วนทุกด้านเตรียมยื่นเอกสารตามข้อกำหนดของ NB-Notified Body
- เมื่อจัดทำ Technical Documentation File และ ตรวจสอบความพร้อมของระบบบริหาร-QMS ผู้ผลิตต้องยื่นขอการรับรองตามกระบวนการ ขั้นตอนจากหน่วยงานให้การรับรองหรือ Notified Body (สามารถดูรายชื่อผู้ให้การรับรองได้จาก website หรือ ชื่อหน่วยงานที่ประกาศโดย EU)
เมื่อผ่านการรับรองแล้ว ผู้ตรวจจะมีแผนการตรวจเป็นรายปีเป็นอย่างน้อย กฎหมาย กฎระเบียบ EU MDR ยังคงติดตาม Update ข้อมูลเครื่องมือแพทย์ที่ผ่านการรับรองเป็นระยะๆ และตาม Risk Classification หรือ สอดคล้องตาม Conformity Assessment และ ข้อกำหนดกฎหมาย MDR กำหนดไว้
จากข้อมูลข้างต้นนี้ เป็นขั้นตอนง่ายๆ โดยคร่าวๆ เพื่อเป็นขั้นพื้นพื้นฐานในการวางแผนดำเนินการ ซึ่งในรายละเอียดส่วนลึก ส่วนหนึ่งขึ้นอยู่กับผลิตภัณฑ์เครื่องมือแพทย์แต่ละประเภท แต่ละ Risk Classification ที่มีกฎระเบียบเฉพาะอีก ต้องศึกษาเพิ่มเติม !!
Our Services
- EU MDR2017/745 Regulations Training
- How to apply to CE Marking
- Clinical Evaluation Training
- Clinical Evaluation Report preparation
- Technical Documentation file preparation
- PMS-Post Market Surveillance training and consultation
แหล่งข้อมูลอ้างอิง
Official Journal of the European Union REGULATION (EU) 2017/745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 5 April 2017