
Technical Documentation หรือ Technical File
แฟ้มที่สำคัญที่ผู้ผลิตไม่ควรละเลยและมองข้าม เพราะแฟ้มเทคนิคของผลิตภัณฑ์นี้ ต้องแสดงความสอดคล้องและตอบข้อกำหนด กฎหมายของ EU เพื่อขออนุญาตนำสินค้าเข้าวางจำหน่ายในสหภาพยุโรป หรือที่รู้จักกันในนาม CE Marking
ซึ่งหากเริ่มต้นด้วยความไม่ถูกต้อง ไม่สอดคล้องตามกฎหมาย จะไม่สามารถได้รับการรับรองและการอนุญาตให้ใช้มาร์ค ตลอดจนนำสินค้าวางจำหน่ายได้ตามต้องการ
ดังนั้นคุณสมบัติของ Technical File ควรต้องมีความชัดเจน สอบกลับได้ ให้รายละเอียดผลิตภัณฑ์เครื่องมือแพทย์ที่ต้องการยื่นขอการรับรองอย่างครบถ้วน มีความทันสมัย เชื่อมโยงและสอดคล้องตามข้อกำหนดกฎหมายระบุ
- Technical Documentation
สำหรับแฟ้มข้อมูลด้านเทคนิคของผลิตภัณฑ์เครื่องมือแพทย์ ในบทความนี้ขออ้างอิงตามกฎหมายเครื่องมือแพทย์ของสหภาพยุโรป หรือที่รู้จักกันดีในชื่อ CE Marking ตามกฎหมาย EU Medical Device Regulation 2017/745 และ EU IVDR2017/746 ใน Technical File เป็นข้อบังคับหนึ่งที่สำคัญ ที่ผู้ผลิตต้องการขอใช้ CE marking แสดงความสอดคล้อง ความปลอดภัยตามกฎหมายสหภาพยุโรป
ในกฎหมาย EU MDR กำหนดใน Annex II ให้มีการจัดทำ Technical Documentation
- Technical File ต้องประกอบด้วยข้อมูลใดบ้าง
ก่อนเริ่มจัดทำ Technical File ที่ปรึกษาขอแนะนำ ผู้ผลิตควรกำหนดขอบเขตผลิตภัณฑ์ให้ชัดเจน เช่นอยู่ในกลุ่มเครื่องมือแพทย์ตามกฎหมายอะไร เป็นเครื่องมือแพทย์กลุ่ม Non-Medical Intended, มีระดับความเสี่ยงใด (Risk Classification) และตรวจสอบ ศึกษาเนื้อหากฎหมาย MDR หรือ CE Mark ให้ครบถ้วนและเข้าใจก่อน เพื่อเป็นประโยชน์ต่อการวางแผน การจัดทำ Technical File ตัวอย่างตาม Flow 1 ข้างล่างนี้ จะทำให้การจัดทำเป็นไปด้วยความรวดเร็ว ง่ายเข้าถึงเนื้อหา และข้อกำหนดกฎหมาย มาตรฐานที่เกี่ยวข้องได้ หากข้อมูลเหล่านี้ครบถ้วนและถูกต้อง จากประสบการณ์ทีมที่ปรึกษาคิวไทม์ จะสามารถดำเนินการจัดทำให้คำปรึกษา Technical File ได้แล้วเสร็จในระยะเวลาไม่นาน
Flow : TDF preparation
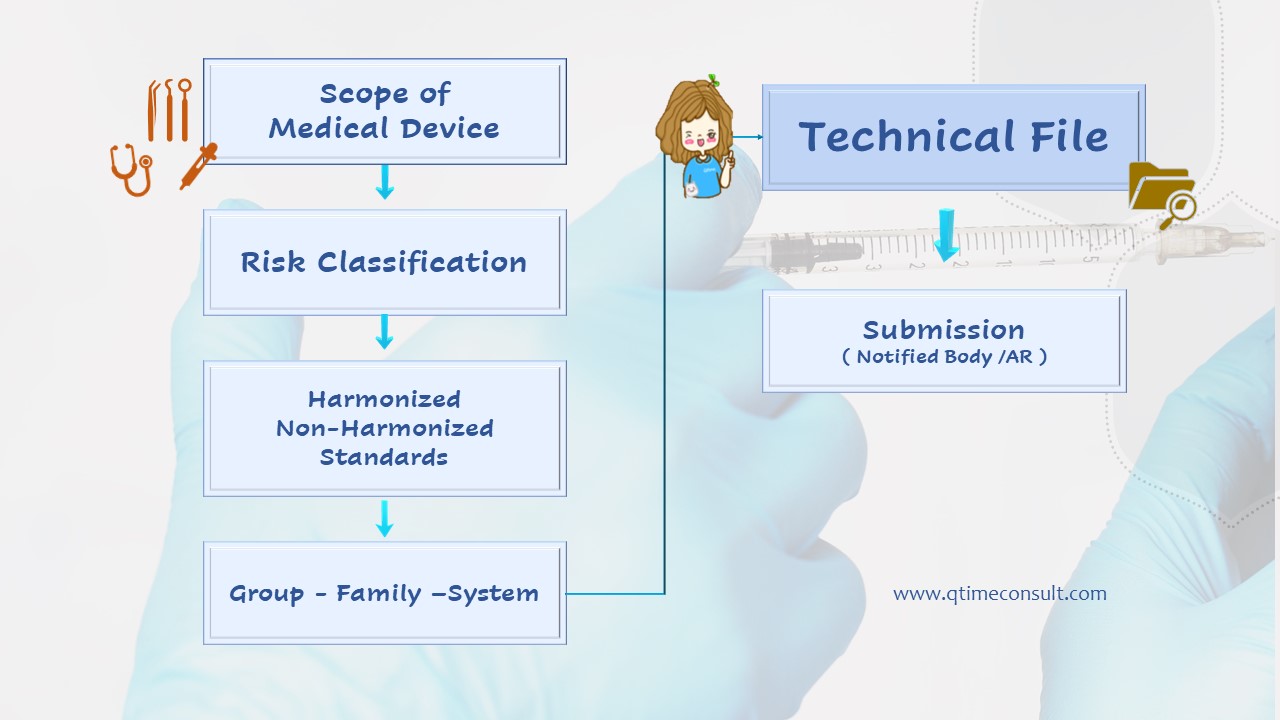
โดยข้อมูลหลัก ๆ แบ่งเป็น 6 หัวข้อใหญ่ด้วยกันคือ
ข้อมูลด้านที่ 1 คือเป็นรายละเอียดเกี่ยวกับผลิตภัณฑ์เครื่องมือแพทย์ที่ต้องการยื่นขอการรับรอง รวมถึงอุปกรณ์ต่อพ่วง อุปกรณ์เสิรม ที่ผลิตภัณฑ์ต้องใช้ร่วมกัน (Device Description and Specification, Accessories) เช่น วัตถุประสงค์และข้อบ่งชี้การใช้งาน รายละเอียดคุณสมบัติ คุณลักษณะของผลิตภัณฑ์ อายุการจัดเก็บ อายุการใช้งาน ปราศจากเชื้อ Basic UDI-DI
ข้อมูลส่วนที่ 2 คือ เอกสารกำกับการใช้งาน หรือ IFU – Instruction for use และ ฉลากหรือ Labeling เนื้อหาการใช้งานเครื่องมือแพทย์ผู้ผลิต ควรต้องตรวจให้ครบ ตามข้อกำหนดกฎหมายกำหนด มาตรฐานกำหนด และการอ้างอิง คำเตือน ข้อควรระมัดระวัง ข้อห้ามในการใช้งาน ความไม่พึงประสงค์ที่อาจจะเกิดขึ้น
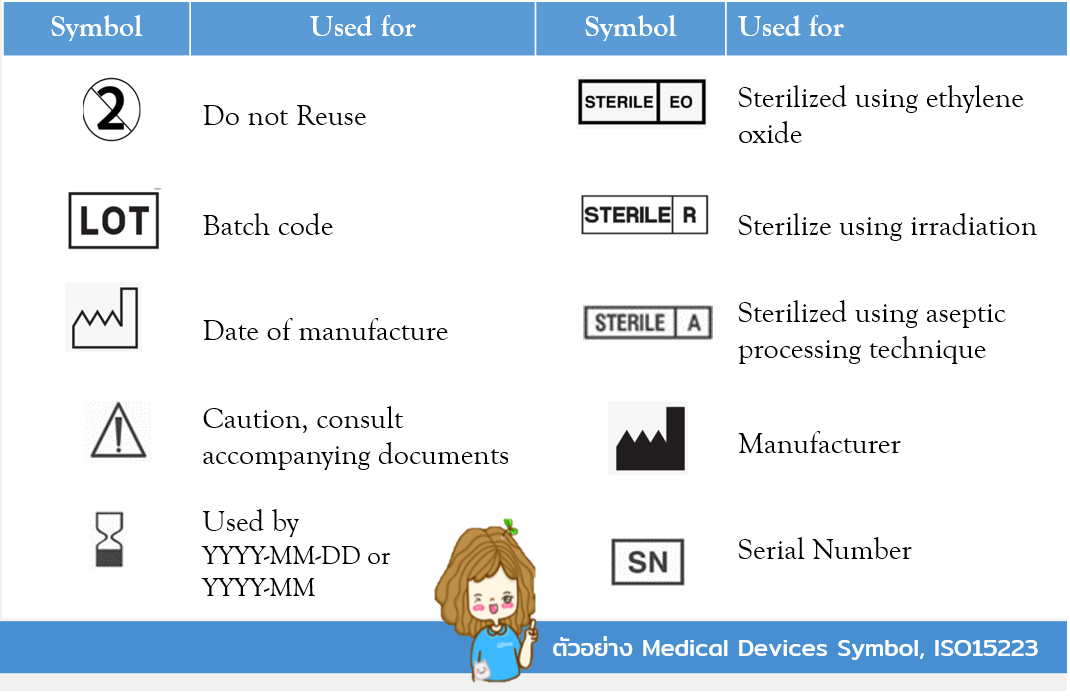
การออกแบบฉลากและการใช้สัญลักษณ์ (Medical Symbol) สอดคล้องตาม ISO15223 Medical Devices-Symbols to be used with medical device labels, labelling and information to be supplied หากเป็นเครื่องมือแพทย์ที่มีกำหนดเฉพาะเจาะจง ควรต้องนำข้อกำหนดนั้นมาใช้ประกอบการจัดทำฉลากด้วย เช่น ข้อกำหนดระบุถึงการจัดเก็บผลิตภัณฑ์ ห้ามแช่แข็ง เมื่อเปิดใช้แล้วต้องใช้ให้หมดภายใน 24 ชั่วโมง เป็นต้น ที่ปรึกษาคิวไทม์ไม่แนะนำเป็นอย่างยิ่งในการสำเนาสัญลักษณ์และข้อความจากผลิตภัณฑ์บริษัทอื่น เพราะแต่ละผลิตภัณฑ์อาจมีความจำเพาะ คุณสมบัติผลิตภัณฑ์อาจไม่เหมือนกัน ดังนั้นฉลากจึงมีคุณสมบัติที่ต้องให้ข้อมูลของผลิตภัณฑ์ได้ไม่สับสน ลดความเสี่ยง ใช้งานได้อย่างเต็มประสิทธิภาพและความปลอดภัย
ตัวอย่างข้อมูลเบื้องต้นในฉลาก เช่น
- ชื่อที่อยู่ผู้ผลิต (Manufacturer information)
- ชื่อและที่อยู่ของ A.R.
- CE Marking Logo
- Sterile บ่งชี้เป็นผลิตภัณฑ์ปราศจากเชื้อด้วยวิธีใด
- Single use (ถ้าเป็นผลิตภัณฑ์ใช้ครั้งเดียว)
- ชื่อผลิตภัณฑ์ รุ่น
- วัตถุประสงค์การใช้
ตัวอย่างสัญลักษณ์ ISO15223
ข้อมูลส่วนที่ 3 การออกแบบ และข้อมูลของผู้ผลิต (DESIGN AND MANUFACTURING INFORMATION) บทสรุปการออกแบบของผลิตภัณฑ์ แบบ รุ่น การตรวจสอบ (Final Inspection requirements) และรายละเอียดของผู้ผลิต (Company sites) ผู้ขาย (Supplier) และผู้รับเหมาช่วง (Sub-contractors)
ข้อมูลส่วนที่ 4 การให้ข้อมูลตามข้อกำหนดของ GSPR -GENERAL SAFETY AND PERFORMANCE REQUIREMENTS ผู้ผลิตต้องนำ Checklist หรือแบบสอบถามนี้มา ใส่ข้อมูลที่เกี่ยวข้อง บ่งชี้ที่เกี่ยวข้องกับผลิตภัณฑ์ อ้างอิงมาตรฐาน (Harmonized standards and Common specification) และผลลัพธ์ในแต่ละเรื่องของ GSPR การกรอกข้อมูลในแบบ checklist หรือ แบบสอบถามนี้มีความสำคัญ เนื่องจากในแต่ละข้อของ GSPR ต้องชี้แจงแสดงให้เห็นความสอดคล้องและไม่เกี่ยวข้องกับข้อกำหนดนั้นอย่างไร
ข้อมูลส่วนที่ 5 เรื่องการวิเคราะห์ความเสี่ยงและประโยชน์-ความเสี่ยง รวมถึงการบริหารความเสี่ยง (BENEFIT-RISK ANALYSIS AND RISK MANAGEMENT) การวิเคราะห์ความเสี่ยงใช้แนวทาง Guidelines : ISO 14971 Risk Management for Medical Device ครอบคลุม Lifecycle ของข้อกำหนด และผลิตภัณฑ์ การกล่าวเคลมในเรื่องประโยชน์และความเสี่ยงโดยข้อมูลและหลักฐานด้าน In-vitro test และ In-vivo test.
ข้อมูลส่วนที่ 6 ข้อมูลด้านการทดสอบผลิตภัณฑ์และการรับรองความถูกต้องของผลิตภัณฑ์ ( VERIFICATION AND VALIDATION : Test Report ในหัวข้อต่าง ๆ คือ
- Electrical Safety and EMC Test
- Biological Test
- Biocompatibility test
- Chemical Characterization and Test
- Aging and Stability test/Study
- Performance test
- Physical Test
- Usability test
และข้อมูลผลทางการด้านคลินิก (Clinical Evaluation หรือ Clinical Investigation ) และการเตรียมทำแผนการติดตามหลังผลิตภัณฑ์วางจำหน่าย (Post Market Surveillance Plan) และการติดตามทางด้านคลินิก (Post Market Clinical Follow up -PMCF)
ในกรณีที่เป็นผลิตภัณฑ์เครื่องมือแพทย์ Class III เช่นใช้วัสดุหรือวัตถุดิบจากมนุษย์หรือจากสัตว์ (Human or Animal origin) ใช้ร่วมกับยา (Drug) ผู้ผลิตต้องเตรียมข้อมูลเพิ่มเติมในเรื่องวัตถุดิบเหล่านี้ที่นำมาใช้
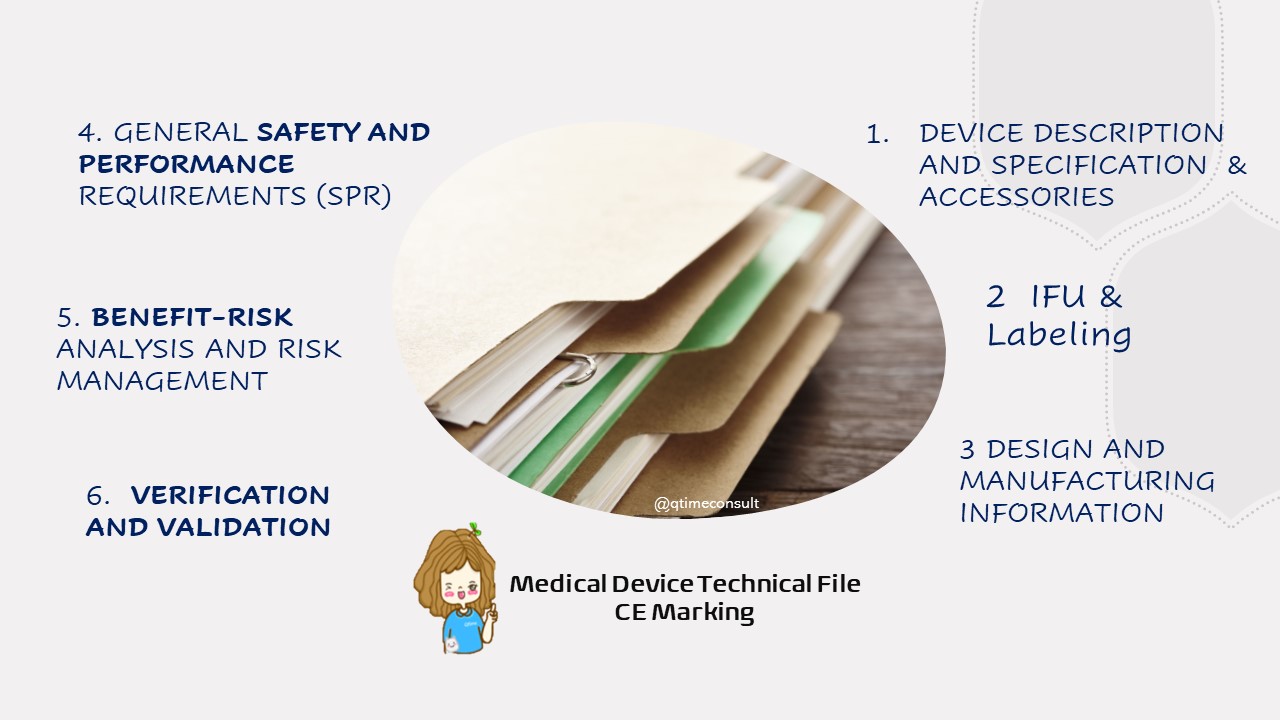
เมื่อจัดทำ Technical File ข้อมูลครบถ้วนแล้ว ผู้ผลิตสามารถจัดส่งให้กับ Notified Body หรือ EC representative เพื่อตรวจสอบรับรองตามความสอดคล้องต่อไป (Conformity Assessment)
- การประเมินความสอดคล้องของ Technical File
ใน Annex IX การจัดทำ Technical File ต้องจัดทำทุก Risk Classification คือ Class I, Class IIa, Class IIb, และ Class III และผู้ตรวจสอบ Technical file โดย NB-Notified Body สำหรับผลิตภัณฑ์เครื่องมือแพทย์ Class IIa, Class IIb, และ Class III
ขั้นตอนการยื่นของการรับรอง CE Marking for Medical Device
ตัวอย่าง Content of Technical Documentation
การใช้บริการของที่ปรึกษา
- Training : EU MDR2017/745 EU MDR2017/745 ข้อกำหนดกฎหมายและการจัดทำ
- Consulting : ครอบคลุมเนื้อหาตามกฎหมาย MDR2017/745
- Technical File : การจัดทำ Technical File
แหล่งที่มาข้อมูล
- Regulation (EU)2017/745 of the European Parliament and of the council of 5 April 2017