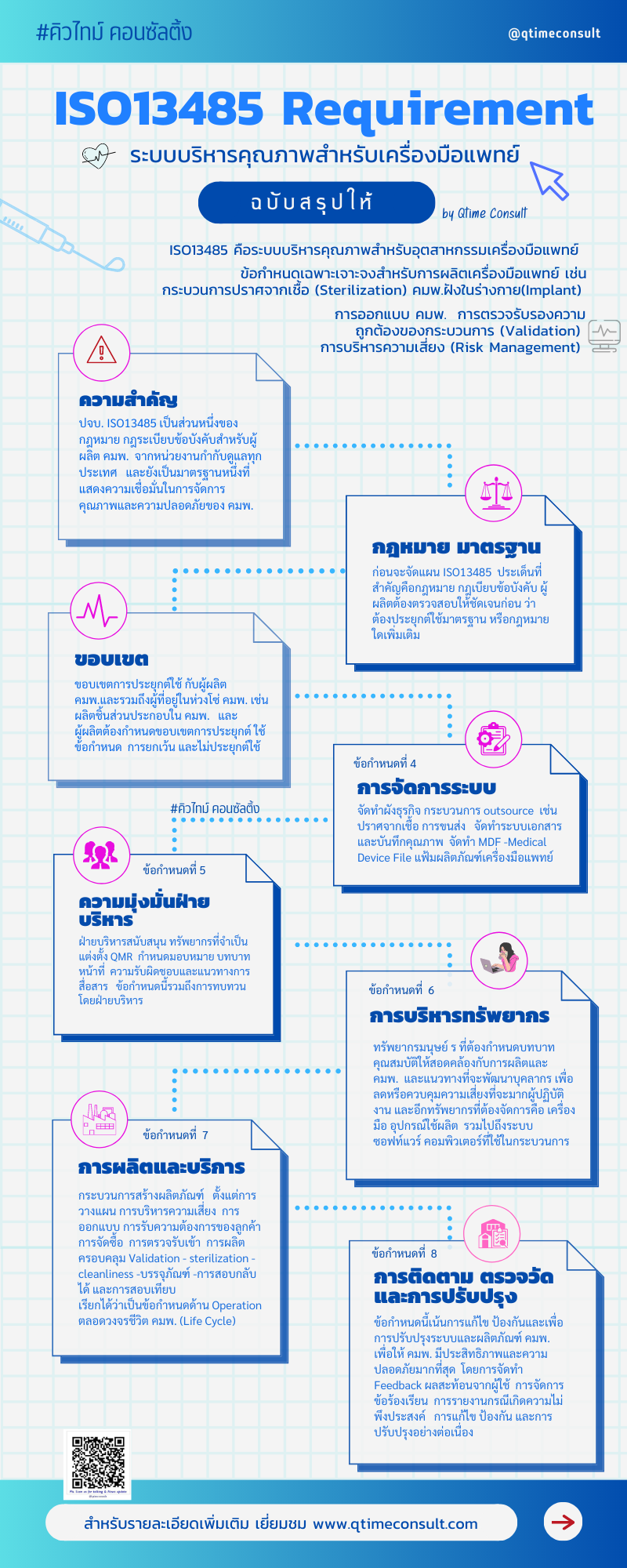
ISO13485 สรุปการประยุกต์ใช้ข้อกำหนด
มาถึงวันนี้ ทุกผู้ผลิตผลิตภัณฑ์เครื่องมือแพทย์ ได้รู้จัก ได้ประยุกต์ใช้ และได้ขอการรับรองจาก Certified Body (CB) กันเกือบทุกผู้ผลิตแล้ว ดังนั้นในการชวนสนทนากันเรื่อง ISO13485 ครั้งนี้ที่ปรึกษา Q Time consult จึงขอนำมา ISO13485
สรุปจากประสบการณ์ของที่ปรึกษา ไม่ว่าจะมาจากการอบรมหรือให้คำปรึกษาที่ผ่านมา
ในแต่ละครั้งและแต่ละสถานประกอบการจะพบเจอปัญหาอุปสรรคที่แตกต่างกันไป ในบางครั้งผู้ผลิต ผลิตภัณฑ์ในกลุ่มเดียวกันแต่ก็พบประเด็นปัญหาไม่เหมือนกัน แต่จะเป็นปัญหาอุปสรรคใดบ้างในแต่ละข้อกำหนด จะนำเสนอเรียบเรียงตามข้อกำหนดข้อ 4 - ข้อกำหนดที่ 8 และแชร์ประสบการณ์ที่ปรึกษา

แต่ก่อนเริ่มเข้าข้อกำหนด ISO13485 ขอทบทวนเรื่องขอบเขตและการประยุกต์ใช้กันอีกสักครั้ง โดย ISO13485 มาตรฐานสากลที่ได้รับการยอมรับทั่วโลก สามารถประยุกต์ใช้ได้กับผู้ผลิต ผู้นำเข้า ผู้จัดจำหน่าย ผู้ให้บริการเก็บรักษาหรือ warehouse ผู้ให้บริการด้านเครื่องมือแพทย์และขอบเขตอื่นๆ ในประเด็นนี้จะแตกต่างจาก GMP Medical Device ที่เน้นที่ผู้ผลิตเครื่องมือแพทย์เป็นผู้ประยุกต์ใช้และขอการรับรองจากหน่วยงานกำกับดูแล ไม่เหมาะกับผู้ผลิตชิ้นส่วนหรือผลิตภัณฑ์ที่ไม่เป็นไปตามวัตถุประสงค์การใช้งานเครื่องมือแพทย์
หลังจากได้ขอบเขตการประยุกต์ใช้แล้ว เริ่มเข้าสู่การทบทวนข้อกำหนด
ข้อกำหนดที่ 4
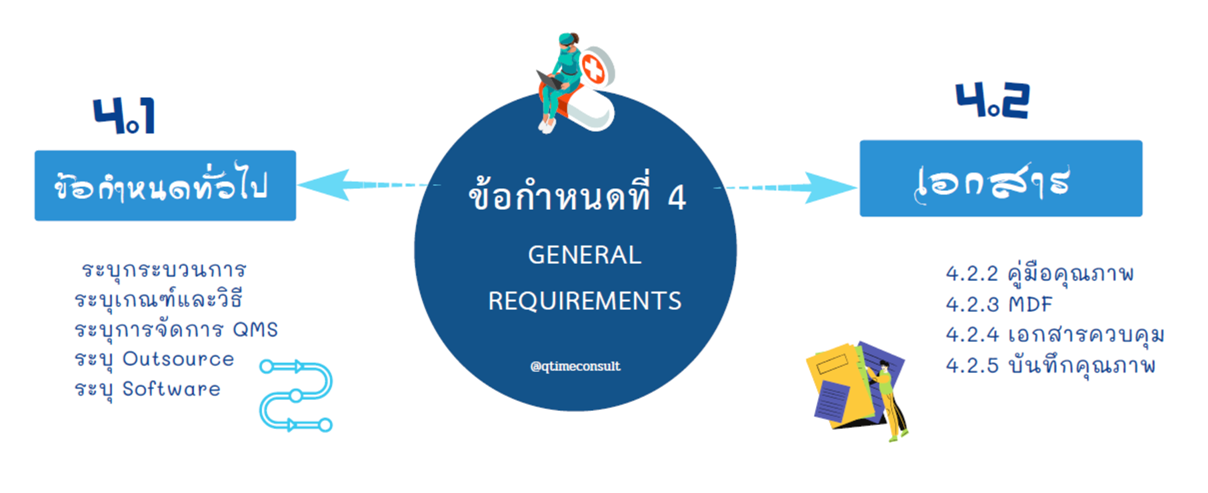
ข้อกำหนดที่ว่าด้วยการเตรียม ตั้งต้นระบบและการจัดการ โดยการจัดระเบียบต้องเขียนผังธุรกิจ (Business Flow) รวมถึงกิจกรรมการผลิต เครื่องมือแพทย์มีหลากหลายลักษณะและวัตถุประสงค์การใช้งาน เงื่อนไขการใช้งานเป็นปัจจัยสำคัญหนึ่งในการกำหนดกระบวนการผลิตและบริการ ถ้าจำเป็นต้องมีผู้รับเหมาช่วง เช่นกระบวนการชุบ กระบวนการปราศจากเชื้อ ผู้ผลิตต้องระบุผู้รับเหมาช่วง (Outsource Process) ให้ชัดเจน ซึ่งหากกำหนดหรือระบุไม่ได้จะมีความล่าช้าในการจัดการ การระบุกระบวนการผู้รับเหมาช่วงที่ชัดเจนว่าเป็นกระบวนการใดยังช่วยให้เราได้จัดการบริหารความเสี่ยงของ Outsource ได้ถูกต้องเพิ่มประสิทธิภาพให้กระบวนการอีกด้วย
ในข้อกำหนดข้อที่ 4 นี้ยังกำหนดให้ จัดทำเอกสารคือ จัดทำคู่มือคุณภาพ(Quality Manual) การจัดทำแฟ้มผลิตภัณฑ์เครื่องมือแพทย์ (Medical Device File-MDF) ซึ่งไม่ใช่แฟ้ม Technical Document file และไม่ใช่ CSDT File ซึ่งเป็นแฟ้มผลิตภัณฑ์ที่ต้องยื่นขอขึ้นทะเบียนผลิตภัณฑ์กับหน่วยงานกำกับดูแล ดังนั้นขอบเขตของ MDF คือรายละเอียดผลิตภัณฑ์ที่ต้องการผลิต ระบุประเภท รุ่น เป็นต้น ผู้ผลิตต้องกำหนดระบบเอกสาร ทั้งเอกสารควบคุม (Document Control -DC) และบันทึกคุณภาพ (Record control) ระบบการจัดเก็บบันทึกคุณภาพต้องกำหนดให้สอดคล้องทั้งกฎระเบียบข้อบังคับ หรือจากมาตรฐานที่เกี่ยวข้อง และจัดเก็บตามอายุการใช้งานและอายุจัดเก็บของผลิตภัณฑ์ หลายๆ ผู้ผลิตกำหนดตามนโยบายของบริษัท เช่น นโยบายจากบริษัทแม่กำหนดให้จัดเก็บ 50 ปี ก็สามารถทำได้เช่นกัน
ข้อกำหนดที่ 5
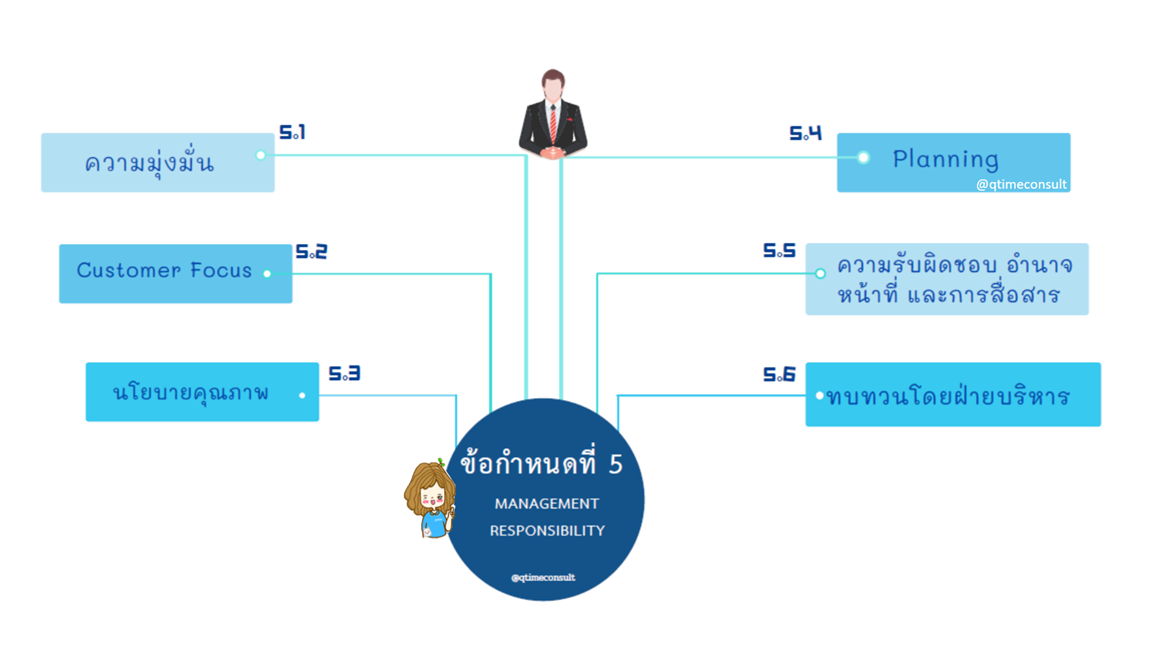
ความรับผิดชอบโดยฝ่ายบริหาร โดยภาพรวมข้อกำหนดเรื่องนี้ของ ISO13485 มีแนวทางเช่นเดียวกับ ISO9001 แต่มีข้อกำหนดบางเรื่องที่แตกต่าง เช่น QMR การแต่งตั้งตัวแทนฝ่ายบริหาร ผู้บริหารแสดงความมุ่งมั่นให้นโยบาย มอบหมายและสนับสนุนทรัพยากรทั้งในเรื่องบุคลากร การแต่งตั้งและจัดทำโครงสร้างผังองค์กร กำหนดบทบาทหน้าที่ การสนับสนุนอุปกรณ์เครื่องจักร และสถานที่โครงสร้างพื้นฐาน เพื่อให้การปฏิบัติงานเกิดประสิทธิภาพ และมีการประชุมทบทวนเป็นระยะๆ โดยภาพรวมผู้ประยุกต์ใช้ สามารถบริหารจัดการในข้อกำหนดนี้ได้ ส่วนหนึ่งอาจมาจากผู้บริหารของทุกองค์กรมุ่งมั่นผลิตเครื่องมือแพทย์ที่มีคุณภาพและความปลอดภัยอยู่แล้ว และมีบางองค์กรที่มีการแต่งตั้ง QMR ตัวแทนฝ่ายบริหารมากกว่า 1 ท่าน ในรูปแบบนี้จัดว่าไม่สอดคล้องข้อกำหนดหรือไม่ ที่ปรึกษาคิวไทม์ ต้องแนะนำให้ทบทวนบทบาทหน้าที่ให้ชัดเจน หากมี 2 ท่าน แต่บทบาทหน้าที่ไม่มีผู้ปฏิบัติงานจริง การกำหนดรูปแบบนี้จะไม่เกิดประโยชน์ต่อการบริหารระบบคุณภาพ เพิ่มความสับสนและความขัดแย้ง
ข้อกำหนดที่ 6
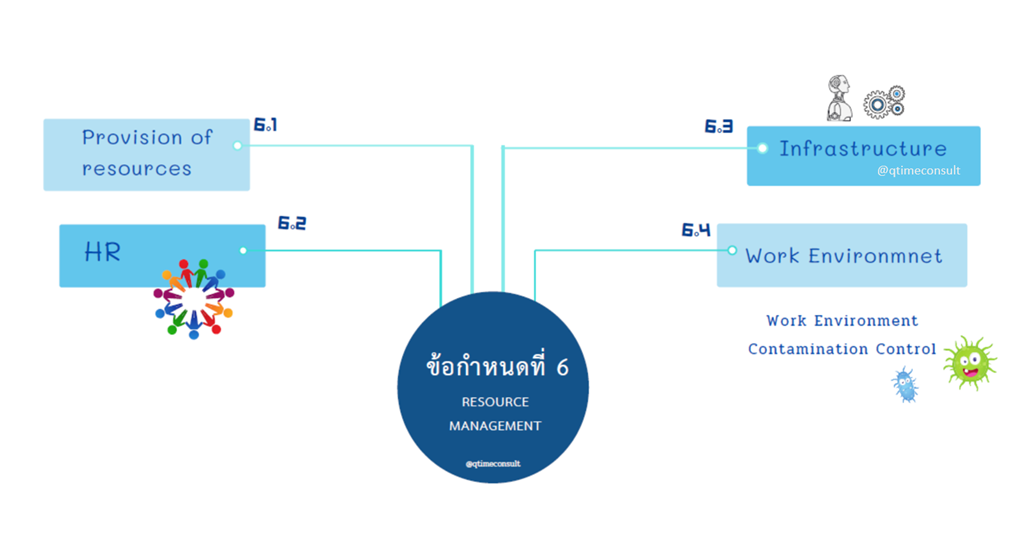
เรื่องบริหารทรัพยากร เป็นข้อกำหนดหนึ่งที่มีความแตกต่างจาก ISO9001 ในเรื่องการไม่ประยุกต์ใช้ (NA-Non applicable) ข้อกำหนดนี้ได้ ทั้งนี้ขึ้นอยู่กับเงื่อนไขผลิตภัณฑ์เครื่องมือแพทย์ เช่น กลุ่มผลิตภัณฑ์ปราศจากเชื้อ ที่การผลิตต้องสะอาดและควบคุมการปนเปื้อนได้ ห้องคลีนรูมจึงเป็นเงื่อนไขหนึ่งที่อยู่การผลิตผลิตภัณฑ์ปราศจากเชื้อ (Sterilization) หลายๆ ท่านเคยสอบถามที่ปรึกษาว่า ไม่ต้องสร้างห้องคลีนรูมได้หรือไม่ ที่ปรึกษาแนะนำว่าควรต้องเป็นคลีนรูม การบริหารจัดการจะจัดการง่ายและเกิดความมั่นใจต่อผู้บริโภคและลดความเสี่ยงจากกระบวนการผลิตได้ด้วย ส่วนอุปกรณ์เครื่องจักรที่ใช้ประกอบการผลิตจัดทำรายการและแผนบำรุงรักษา ระบุผู้รับเหมาช่วง คุณสมบัติใบรับรองหรือมาตรฐานที่เกี่ยวข้องให้ชัดเจน ทั้งนี้เพื่อให้มั่นใจว่า Outsource ที่เลือกใช้บริการสอดคล้องถูกต้อง ตามกระบวนการผลิตเครื่องมือแพทย์ การควบคุมสภาพแวดล้อมในการผลิต ผู้ผลิตสามารถประยุกต์ใช้ได้ทั้ง 5ส และ GMP รวมถึงการควบคุมการผลิตในพื้นที่คลีนรูม (Cleanroom) การเข้าให้คำปรึกษาของที่ปรึกษาคิวไทม์ให้ความสำคัญเรื่องสภาพแวดล้อมเป็นลำดับต้น ๆ เพราะหากสภาพแวดล้อมถูกต้อง สอดคล้องและควบคุมได้ กระบวนการอื่นๆ จะบริหารจัดการได้ไม่ยาก
ข้อกำหนดข้อ 7
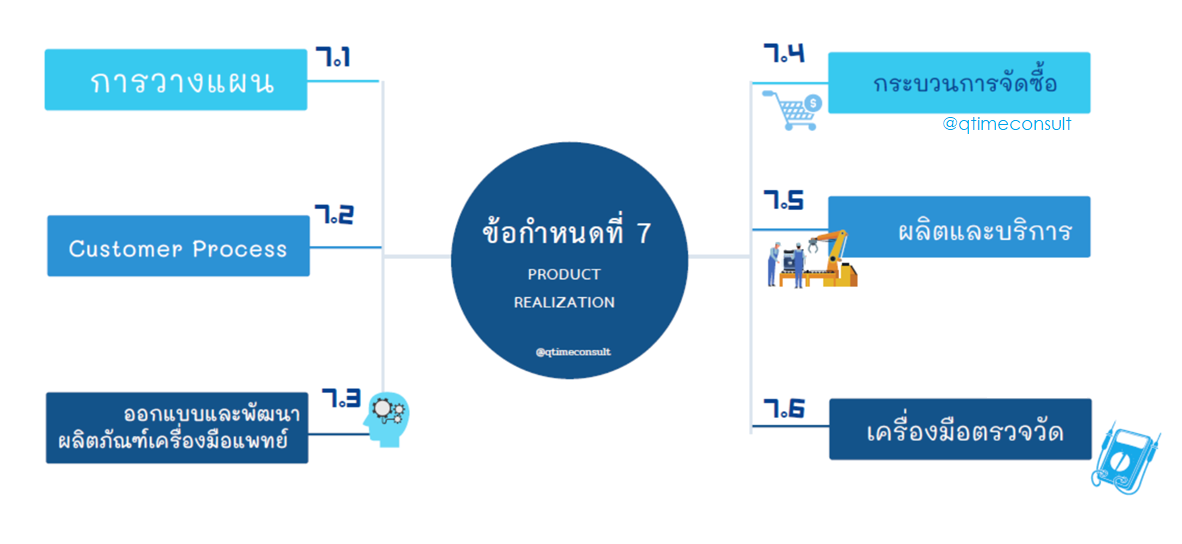
กระบวนการสร้างความเชื่อมั่นให้กับผลิตภัณฑ์หรือกระบวนการสร้างผลิตภัณฑ์ (Product Realization) เป็นกระบวนการหลักของทุกองค์กรไม่ว่าขอบเขต เป็นผู้ผลิตหรือผู้ให้บริการประยุกต์ใช้ตามกิจกรรมการขององค์กร มีข้อกำหนดที่สามารถยกเว้น (Exclusion) ได้คือ กระบวนการออกแบบและพัฒนาผลิตภัณฑ์ และมีข้อกำหนดที่ไม่ประยุกต์ใช้ได้ (Non-Applicable) ตามกระบวนการที่กำหนด เช่น ไม่ประยุกต์ใช้ข้อกำหนดเครื่องกำหนดเครื่องมือแพทย์ปราศจากเชื้อ (Sterilization) ไม่ประยุกต์ใช้ข้อกำหนดการบริการและการติดตั้ง
ในที่นี่ ที่ปรึกษาขอนำข้อกำหนดบางข้อที่สำคัญ นำมาเล่าสู่กันฟัง บางข้อกำหนดที่เป็นกระบวนการคุ้นเคยอยู่แล้ว เช่นการจัดซื้อ ที่ปรึกษาไม่ได้นำมากล่าวถึงในที่นี้
ข้อกำหนด 7.1 ข้อกำหนดนี้ผู้ผลิตจะพบกับเรื่องการบริหารความเสี่ยง (Risk Management) ตามแนวทาง ISO14971 เป็นข้อกำหนดที่ละเว้นไม่ได้และต้องประเมินความเสี่ยงให้ครบกระบวนการ กิจกรรมที่ดำเนินการ
ข้อกำหนด 7.3 การออกแบบและพัฒนาผลิตภัณฑ์เครื่องมือแพทย์ ซึ่งมีข้อกำหนดเรียงตามขั้นตอนการออกแบบ หากขอบเขตมีการจัดทำกระบวนการออกแบบ สามารถเขียน SOP หรือขั้นตอนปฏิบัติงานเรียงตามข้อกำหนดนี้ได้ จะทำให้ไม่สับสนและง่ายต่อการทวนสอบด้วย
ข้อกำหนด 7.5 ข้อกำหนดที่สำคัญและจัดเป็นกระบวนการผลิตและการบริการ ข้อกำหนดนี้ หากผู้ผลิตกำหนดขอบเขตชัดเจนแล้ว จะไม่ยุ่งยากและไม่สับสนในการประยุกต์ใช้ เช่นการติดตั้งและบริการ การจัดการกระบวนการปราศจากเชื้อ เป็นขั้นตอนการผลิตที่ในแต่ละกระบวนการอาจต้องใช้เวลาในการดำเนินการ เช่นการตั้งต้นการผลิต
ข้อกำหนด 7.5.6 Process Validation การตรวจรับรองความถูกต้องของกระบวนการ โดยเฉพาะการผลิต ผู้ผลิตต้องเขียนขั้นตอนการปฏิบัติงาน (SOP) จัดทำแผนและจัดทำรายงานครอบคลุมทุกขั้นตอนคือ IQ-Installation Qualification, OQ-Operation Qualification และ PQ-Performance Qualification
{kind=link}
ข้อกำหนดที่ 8
การตรวจวัด วิเคราะห์และการปรับปรุง
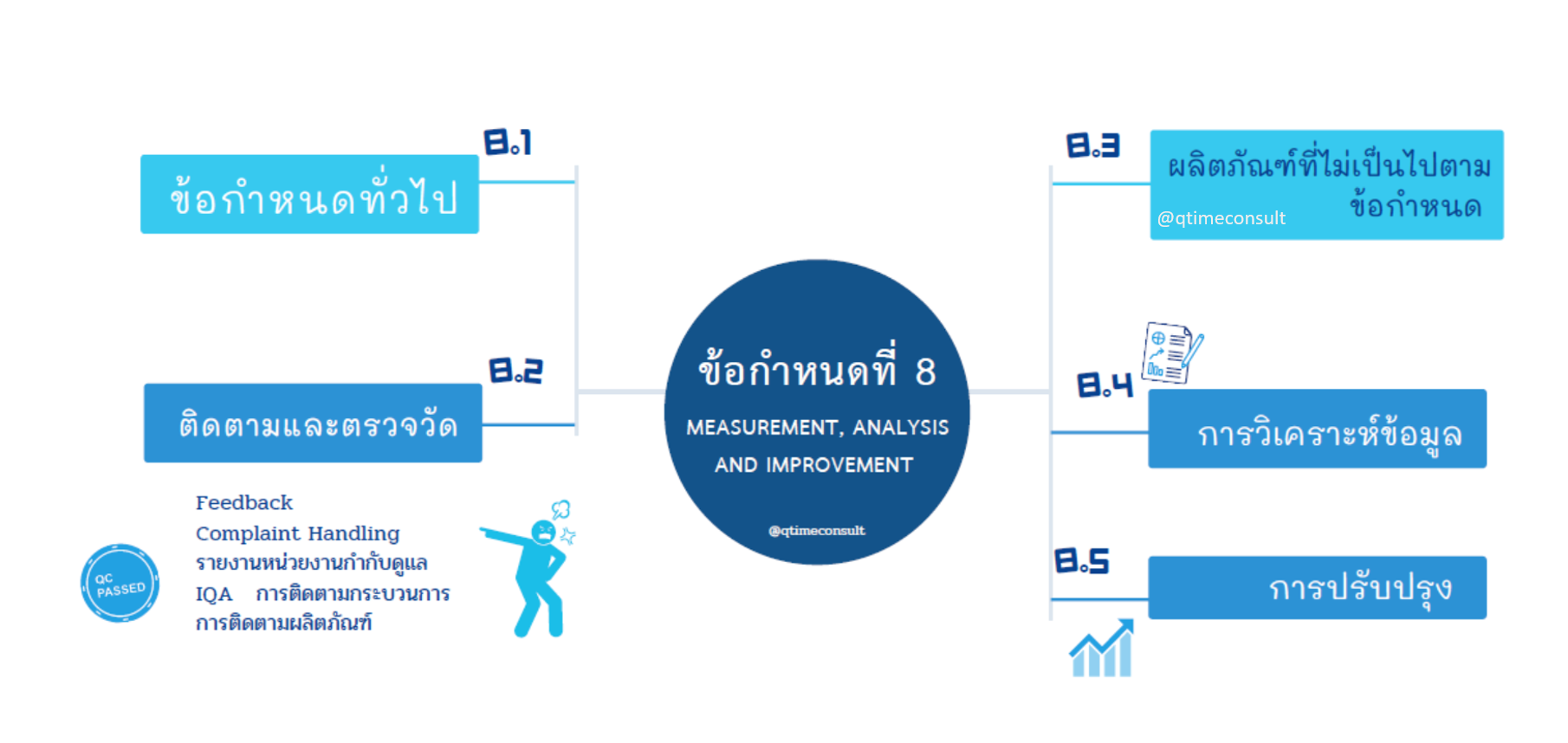
วัตถุประสงค์ข้อกำหนดในข้อที่ 8 นี้ เป็นการเก็บรวบรวม วิเคราะห์ข้อมูลที่มาจากหลากหลายแหล่ง คือ ข้อมูลจากผลสะท้อนกลับหรือ Feedback ข้อร้องเรียน ข้อมูลจากวัตถุประสงค์และเป้าหมาย รายงานที่เกี่ยวข้อง และข้อมูลจากกระบวนการผลิต เพื่อตรวจวัดประสิทธิผลและเพื่อการปรับปรุง การตรวจวัดและวิเคราะห์แบ่งเป็น 2 ส่วนคือ การติดตามวัดกระบวนการระบบบริหารคุณภาพ และการตรวจวัดผลิตภัณฑ์หรือการควบคุมคุณภาพของผลิตภัณฑ์ (Quality Control) ในกระบวนการผลิตหากมีการทำซ้ำหรือ rework ควรต้องระบุประเด็นและวิธีการ rework ให้ชัดเจนด้วยเช่นกัน เพื่อยืนยันและมั่นใจได้ว่า จะมีผลิตภัณฑ์ที่มีคุณภาพและความปลอดภัยตามกำหนดไว้
นอกจากนี้ ผู้ผลิตเครื่องมือแพทย์ ต้องศึกษาและเข้าใจหากเกิดอุบัติเหตุหรือความไม่พึงประสงค์ (Adverse Event ) กับผู้ใช้หรือผู้ป่วย ต้องมีวิธีการดำเนินการอย่างรวดเร็วและถูกต้องตามกฎระเบียบข้อบังคับกำหนดไว้
จากการสรุปข้อกำหนดโดยภาพรวมข้างต้นนี้ ที่ปรึกษายังมีประเด็นหรือเทคนิคที่สามารถนำมาประยุกต์ใช้ ทั้งนี้ขึ้นอยู่กับผลิตภัณฑ์เครื่องมือแพทย์ และกระบวนการที่ผู้ผลิตกำหนด
การประยุกต์ใช้ข้อกำหนดและระบบบริหาร สามารถปรับเปลี่ยนปรับปรุง ได้อยู่ตลอดเวลา มาจากหลากหลายปัจจัย เช่น การพัฒนาผลิตภัณฑ์ใหม่ การปรับเปลี่ยนกระบวนการผลิต ดังนั้น จึงจำเป็นต้องเฝ้าระวัง กำหนดแนวทางปฏิบัติ เพื่อควบคุมการเปลี่ยนแปลง (Change Management) ทั้งนี้ เครื่องมือแพทย์มีส่วนกับชีวิตผู้ป่วย การเปลี่ยนแปลงใดใด อาจมีผลกระทบ ผู้บริหารจึงต้องให้ความสำคัญ
สุดท้ายนี้ ต้องขออภัยหากไม่ได้กล่าวถึงข้อกำหนดใด หากต้องการข้อมูลเพิ่มเติมสอบถาม หรือต้องการแชร์เรื่องใด ต้องการให้ที่ปรึกษาแนะนำเรื่องใด ทักทายได้ที่ @qtimeconsult
- ข้อมูลเพิ่มเติม ขั้นตอนการให้คำปรึกษาระบบ ISO13485
- บทความ การประยุกต์ใช้ GMP สำหรับเครื่องมือแพทย์
- ISO13485 Requirements (ฉบับสรุปให้)
{kind=link}