CE Marking for Medical Devices
CE Marking for Medical Device เป็นมาร์คแสดงคุณภาพและความปลอดภัย ตามกฎหมาย กฎระเบียบและข้อบังคุบของสหภาพยุโรป ผู้ผลิตหรือเจ้าของผลิตภัณฑ์ต้องการนำเข้าผลิตภัณฑ์วางจำหน่ายในสภาพยุโรปต้องแสดงการรับรอง ตามข้อกำหนดของ CE Mark ก่อนใช้และการนำเข้า
สำหรับเครื่องมือแพทย์ (Medical Device) กฎหมายหรือกฎระเบียบที่ ต้องประยุกต์ใช้ คือ EU MDR2017/745 เป็นกฎหมายที่ได้ update และประกาศใช้ใหม่ มีผลบังคับใช้ ในปี 2020 (แต่เนื่องจากสถานการณ์การแพร่ระบาดของ COVID-19 จึงประกาศเลื่อนใช้ พร้อมด้วยกฎหมายสนับสนุน CE Mark อีกหลายฉบับที่เกี่ยวข้องและประกาศเพิ่มเติม
หากผู้ที่ต้องการประยุกต์ใช้และขอการรับรอง CE Marking for Medical Device ต้องพิจารณารายละเอียดในขั้นต้น คือ
ขั้นตอนการจัดทำและขอการรับรอง CE Marking for Medical Devices
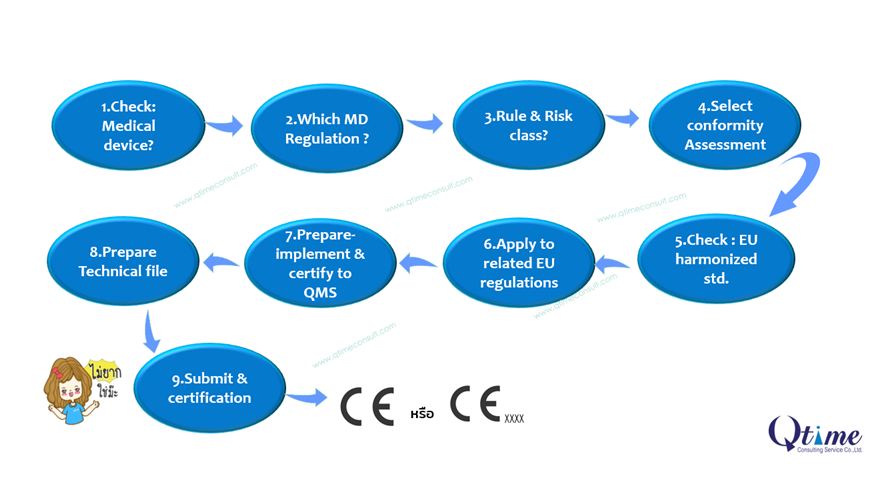
1. Check:Medical Device ? ตรวจสอบว่าเป็นเครื่องมือแพทย์หรือไม่ โดยอ้างอิงความหมาย (Definition) กลุ่มผลิตภัณฑ์ เป็นเครื่องมือแพทย์กลุ่มใด คือ Medical Device (MD) หรือ In-Vitro Diagnostic (IVD) การวินิจฉัย
ความหมาย "Medical Device" (refer to EU MDR2017/745)
‘medical device’ means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
-
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of diseasediagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological or pathological process or state.
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,
and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
The following products shall also be deemed to be medical devices:
-
- devices for the control or support of conception;
- products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
2. What are regulated product? กำหนดกฎหมายที่เกี่ยวข้อง CE Marking for Medical Device คือ EU MDR2017/745 สำหรับ เครื่องมือแพทย์ทั่วไป และ EU IVDR2017/746 สำหรับเครื่องมือแพทย์กลุ่ม IVD หรือเครื่องมือแพทย์วินิจฉัย คัดกรองโรค ทั้งนี้ต้องพิจารณาตามความหมายของเขตของ EU regulation ที่ได้ประกาศด้วย
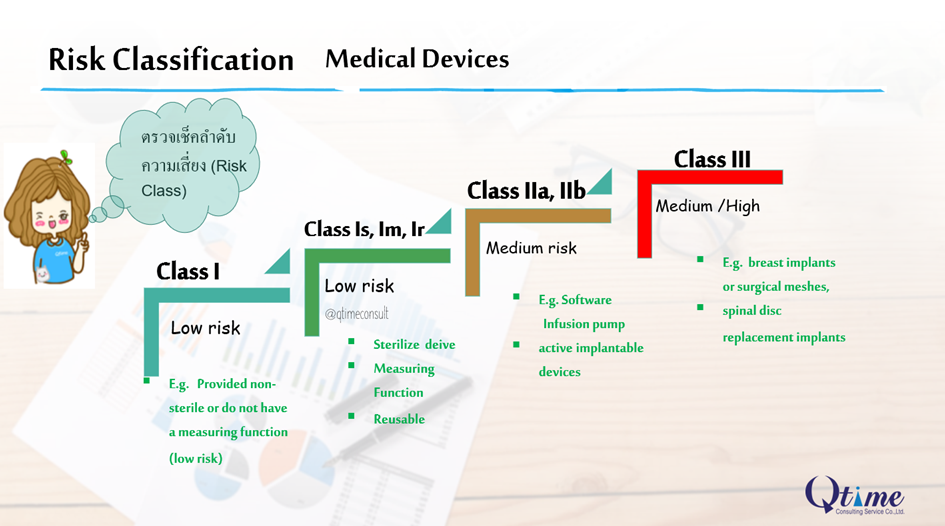
3. Rule and Risk Class? ขั้นตอนต่อมาคือ เมื่อทราบกลุ่มประเภทและกฎหมายที่เกี่ยวข้องแล้ว ตรวจสอบระดับความเสี่ยง เครื่องมือแทพยฺ์ หรือ Risk Classification สำหรับ CE Mark มี 3 ระดับ คือ Class I, IIa, IIb และ III EU MDR2017/745 กำหนดใหม่มี จำนวน 22 rules (click : rule of MD) และ EU IVDR 7 rules เมื่อจัดเข้าเกณฑ์ (rule) แล้ว จะสามารถพิจารณา ระบุระดับความเสี่ยง หรือ Risk Classification ได้
4.select : Conformity Assessment การกำหนดวิธีแสดงการประเมินการรับรองการใช้เครื่องหมาย CE Mark ตามวิธีของ EU MDR จะแบ่งเป็น 2 แนวทางใหญ่ ๆ คือ Class I การรับรองการใช้มาร์คโดยวิธี Self declaration และ Risk class IIa, IIb, III, Is Ir, Im ต้องขอการรับรองผ่าน Notified Body
5. Check : EU harmonized std. and CS std. เลือกมาตรฐานด้านคุณภาพและความปลอดภัยของผลิตภัณฑ์เครื่องมือแพทย์ โดยในขั้นต้นตรวจสอบจาก การประกาศจาก Official Journal of the European Union หากไม่พบประกาศใน Official Journal ผู้ผลิตต้องกำหนดเลือกมาตรฐานเอง เช่นจาก ISO , ASTM หรือตามลำดับขั้นการยอมรับมาตรฐานของ EU ประกาศ
6. Plan and Apply จัดทำแผน เพื่อจะเก็บรวบรวมข้อมูลของผลิตภัณฑ์ ตาม Conformity Assessment ที่เลือกไว้
7. Implement and QMS certification ข้อกำหนดที่สำคัญหนึ่งในเรื่องการประยุกต์ใช้ ISO 13485 หรือระบบ Quality Management System ที่กำหนดให้ ISO13485 เป็นมาตรฐานที่ยอมรับ ดังนั้นบางผู้ผลิต ได้ผ่านการรับรอง ISO9001 หรือ GMP แล้ว แต่ยังคงต้องขอการรับรอง ISO13485 ด้วย
8. Prepare Technical Document File จัดเตรียมแฟ้มเทคนิคของผลิตภัณฑ์เครื่องมือแพทย์ที่ต้องการยื่นขอการรับรอง สอดคล้องตาม Conformity Assessment ที่ได้เลือกหรือกำหนดไว้ หัวข้อหลัก ๆ หรือ Content of Technical Documentation ที่ต้องจัดทำต้องสอดคล้องทั้งผลิตภัณฑ์และ DoC ด้วยเช่นกัน
9. Submit & Certification ขั้นตอนการยื่นขอการรับรอง เบื้องต้นควรต้องขอการรับรอง ISO 13485 ก่อนและยื่นแฟ้ม Technical File ไปยังหน่ยงานที่ยื่นขอการรับรอง
CE Mark คือข้อกำหนดกฎหมายโดยสหภาพยุโรป มีข้อกำหนดอื่นๆ ที่ต้องจัดทำเพิ่มเติม ในรายละเอียดแต่ละขั้นตอน เช่น การแต่งตั้ง EC Representative, EO -Economic Operators, EUDAMED, UDI, PMS, CER เพื่อให้ครบถ้วนทั้งเอกสารที่ต้องการยื่นรับรองการใช้เครื่องหมาย CE Mark และรวมถึงการนำไประยุกต์ใช้กับระบบบริหารคุณภาพ เช่น ISO13485 อย่างต่อเนื่องอีกด้วย
สำหรับบทบาทหน้าที่ของที่ปรึกษา ในการจัดทำ CE Mark, ISO13485 , Technical Documentation File ตลอดจนการยื่นขอการรับรองการใช้ CE mark จะติดตามและแนะนำ step by step
ติดต่อสอบถามขอข้อมูล ได้ที่ @qtimeconsult หรือ ทุกช่องทางการติดต่อ หรือสนใจ CE Marking Training Courses
แหล่งอ้างอิง
EU MDR2017/745 REGULATION (EU) 2017/ 745 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL - of 5 April 2017
EU IVDR2017/746 REGULATION (EU) 2017/ 746 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL - of 5 April 2017 - on in vitro diagnostic medical devices and repealing Directive 98/ 79/ EC and Commission Decision 2010/ 227/ EU (ce-mark.com)